Research
Pleasantries
A wrinkled page, smelling of pickle and sulphur. You have found my publications list, each paper is accompanied by a description of the arcane magics within. They should be appreciable by visiting wizards of any school of magic. If your area of arcana matches mine, mind that this will be rife with generalisations, and be sure to inspect the original tomes for a more rigorous account.
Overview
Currently, all of my publications relate in some way to organic molecular crystals, that is, the state of matter where molecules, made up of mostly carbon, stack together like Lego bricks in order to form a solid. An ice cube, for example, is a molecular crystal of water (though not an organic one). An example of an organic molecular crystal would be a paracetamol tablet, and here is roughly what it looks like (I also included the benzene crystal because I think it looks a little nicer):
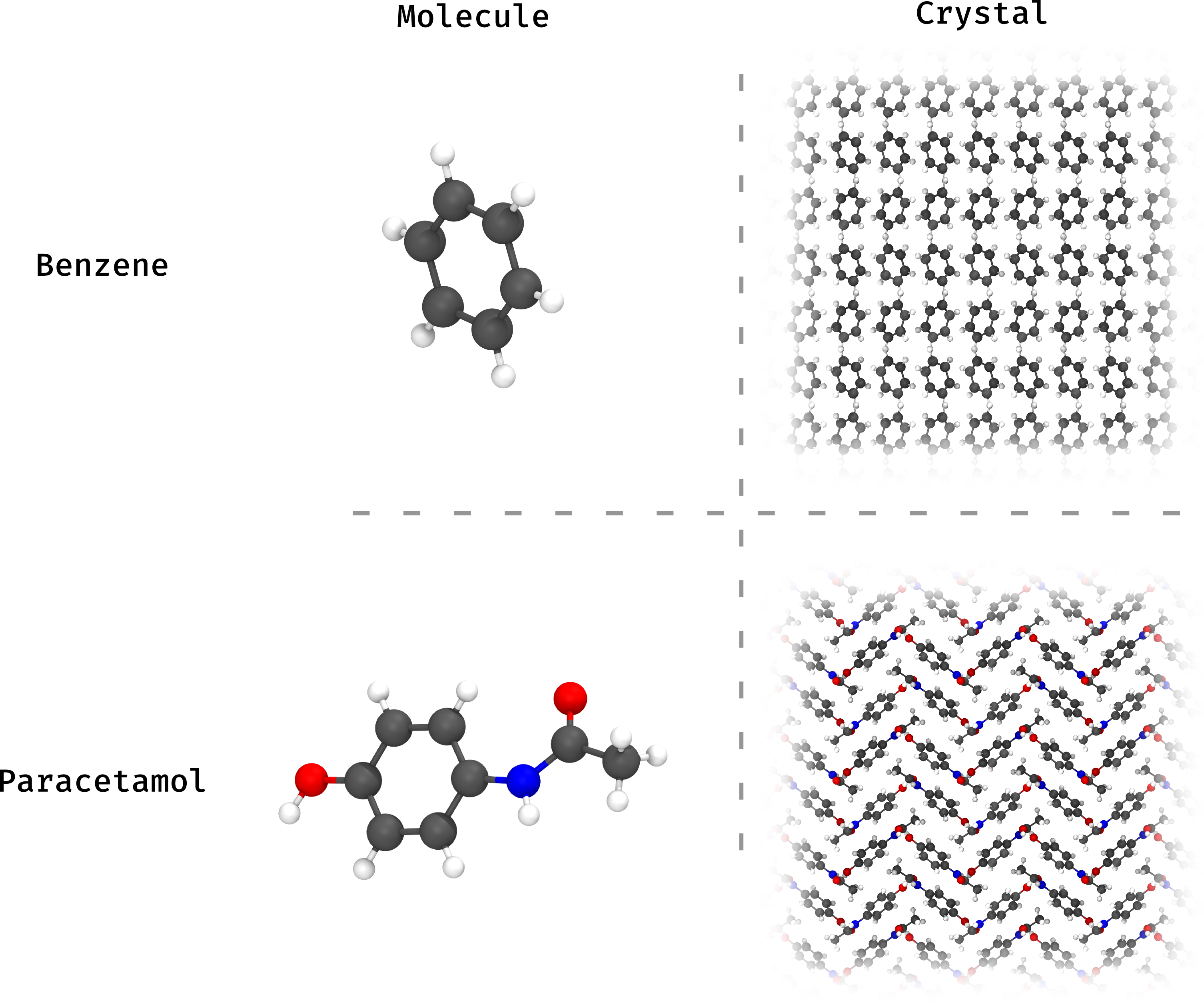
Organic molecular crystals have many applications, and I am in particular interested in their uses which have to do with light. They are used as materials for lasers, solar pannels, electronics, sensors in the bloodstream for medical applications, and LEDs (you might have heard of OLEDs). These materials are light, sometimes flexible, and made up of very common atoms, making them more sustainable than ones which require rare elements.
In order to discover and design them, it is extremely handy to be able to model them on a computer. This way, instead of spending millions of hours and chemicals in a lab trying out different experiments, you spend hundreds of hours and coffee pots in front of a computer, trying out different calculations. Once you have narrowed down some nice candidate, you can send over the recipe to some experimental chemists, who can actually build the thing, and hopefully build some new material.
My role in this picture is to write the programs which model the crystals in question. I design the mathematical tools to model the quantum physics of light interacting with the material, and then translate them into computer programs, which supercomputers are able to understand.
List of Publications
-
How inter-and intramolecular processes dictate aggregation-induced emission in crystals undergoing excited-state proton transfer
Michael Dommett, Miguel Rivera, Rachel Crespo-Otero, J. Phys. Chem. Lett. 2017, 8, 6148-6153
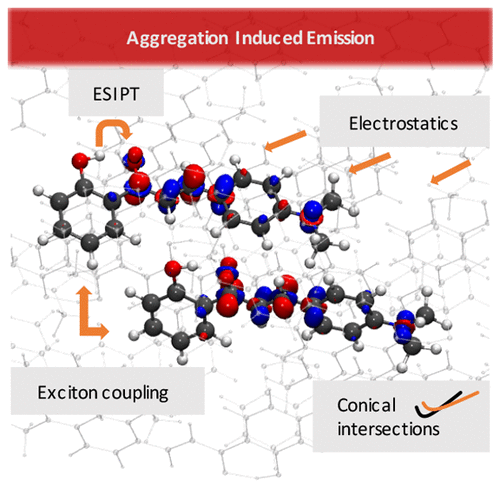
This was an investigation into an unusual property of some organic molecular crystals called Aggregation Induced Emission (AIE). Normally, if molecules emit light when they are dissolved in a solvent (e.g. water), when they are brought together (aggregated) to form a crystal, they stop emitting. In certain cases, the opposite process is true, where a molecule which is dark when dissolved starts emitting as a crystal, thus the name ‘Aggregation Induced Emission’.
We are usually interested in light-emitting crystals rather than solutions for technological applications, since they are solids rather than liquids, therefore being able to predict which dark molecules in solution would become bright in crystal would be very useful. Unfortunately the reasons for AIE are still poorly understood, and this paper makes progress in this direction.
We focused on a family of molecules used in lasers which have a hydrogen atom moving from one part of the molecule to the other when they absorb light. An important point to understand here is that before being able to emit any kind of light, the molecules first need to absorb light, there is never an emission of light coming from nowhere. Thus, whatever determines AIE will have a lot to do with what happens to the molecule after it absorbs light. In this case, the hydrogen moving across the molecule changes a lot about how the atoms move, and can sometimes lead to the material losing the energy from the absorbed photon without emitting any photon in return (through heat). This is why we care about ‘excited state proton transfer’ molecules. ‘Excited state’ means after absorption, and a ‘proton’ is the name of the hydrogen nucleus, therefore the title of the paper refers to crystals made up of molecules which have hydrogens hopping around upon absorbing a photon.
In this family of molecules, some had AIE, while others remained dark in the crystal, despite the difference between the molecules being of only a few atoms. It surfaced that the seemingly small change at the scale of the molecule amplified into a large change in how it stacked together to make a crystal. You can understand it like this: if you were playing Jenga with L-shaped blocks, you wouldn’t be able to stack the tower in the usual way, you would have to come up with a more elaborate architecture. The same was true for these crystals. Therefore there were changes within the molecule itself (intra-) and between the molecules due to the stacking (inter-). These changes were subtly correlated, making it impossible to understand how one affected AIE without the other. The main point of view of the paper was to understand the interplay between these inter- and intramolecular processes, as the title suggests.
This was mostly the work of my groupmate Michael, though I helped in designing and adapting tools to model the ‘excited state’ molecule (read: molecule after absorbing a photon) whilst taking into account the fact that it was within a crystal.
-
ONIOM(QM:QM′) Electrostatic Embedding Schemes for Photochemistry in Molecular Crystals
Miguel Rivera, Michael Dommett, Rachel Crespo-Otero, J. Chem. Theory Comput. 2019, 15, 2504-2516, preprint
This was my first paper as lead author, and ended up being quite the behemoth, being the longest one written during my PhD. In here, we presented a new strategy to model molecules interacting with light in a crystal environment.
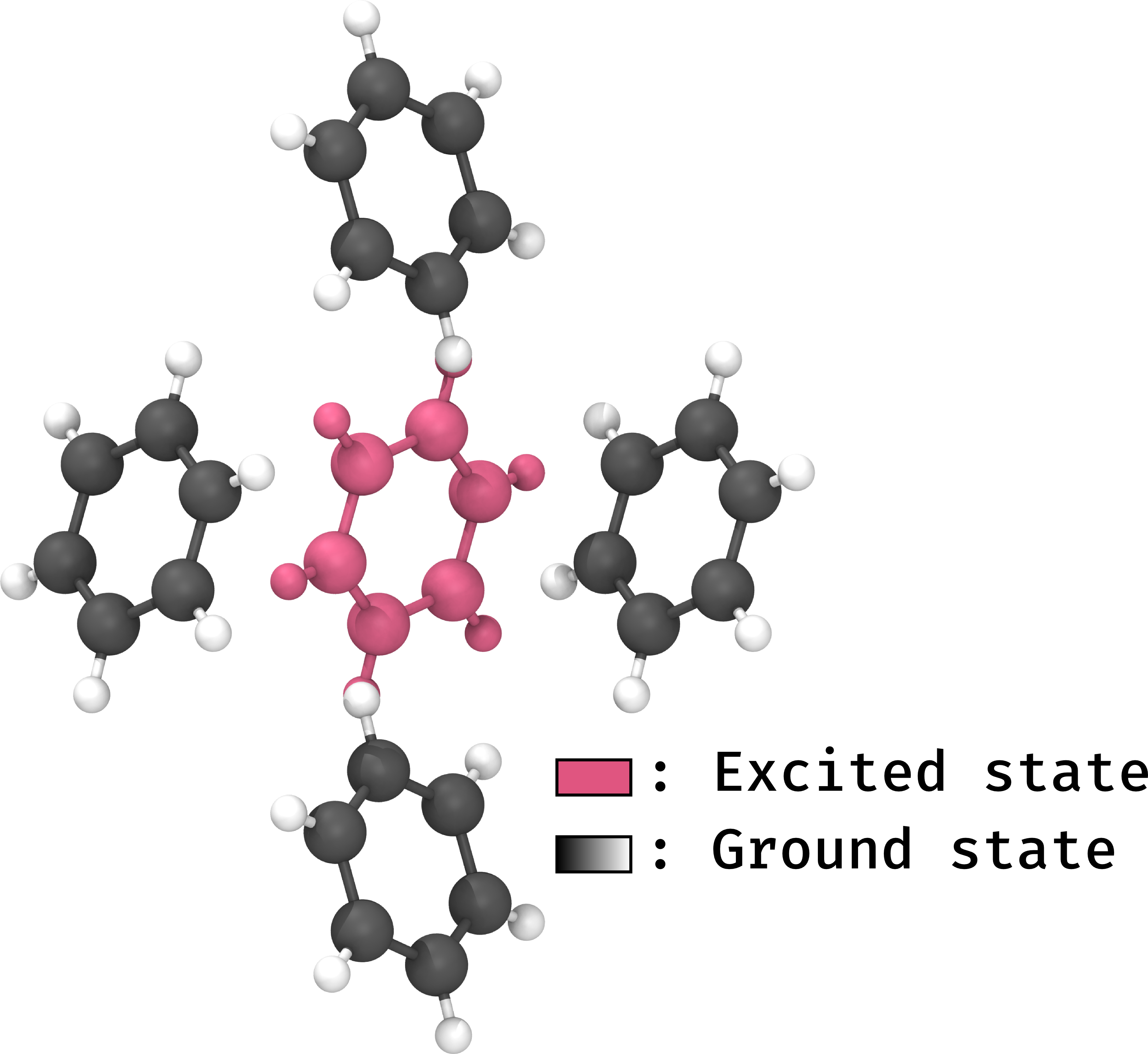
Molecules which have absorbed light are said to be in an ‘excited state’, as opposed to the ‘ground state’. They are much more costly to model since ground states are the lowest possible energy available to the electrons in the molecule. Therefore there is a systematic way of finding a ground state: try a wavefunction (the quantum representation of the electrons), and change it until it reaches its lowest energy. The process is much more complicated for excited states.
Therefore modelling the excited states of a crystal would be impossibly costly, even for a supercomputer, since the amounts of atoms within a crystal are counted in units of 1023. Fortunately for us, organic molecular crystals are not bound together very strongly, which has the consequence that the excited states in these systems stay on one or two molecules, instead of immediately hopping to the whole crystal in one go.
We would therefore like to model one or two (I will use the singular from now on for ease of use) molecules in the excited state. However the fact that they are inside a huge (for our purposes infinite) array of ground state molecules should affect the excited state in some way. In this paper, we developed a way of combining several ground and excited state calculations to model one central molecule in the excited state, surrounded by molecules in the ground state taken from their theoretical positions in the crystal. It looks a bit like this:
This combination of methods is based on a previously designed method called ONIOM (Our own N-layered Integrated Molecular Orbital and Molecular mechanics). The full name is somewhat cryptic, but the acronym is meant to conjure the image of an onion, with layers modelled with different levels of theory (excited state, ground state, or even non-quantum methods). In this case, we care about using quantum methods for both layers of our onion, thus the method is said to be an ONIOM QM:QM’ method where QM stands for Quantum Mechanics, and the apostrophe indicates that it is not the same as the previous QM.
By using ONIOM, the excited state was represented, taking into account all the interactions between it, and its nearest neighbours. However, the interactions between molecules have many components, some of which reach further than just the neighbouring molecules. In particular, electrostatic interactions have a range spanning thousands of atoms, but we are only in a position to model quantum ground states of the order of hundreds. To address this problem, we embedded the onion of excited and ground state molecules in a huge array of placeholder points called ‘point charges’. These accounted for the electrostatic interactions we were after, without having to calculate the wavefunction of thousands of atoms. Additional tweaks were tested out like, using variable amounts of point charges with different values, or making the values of the point charges change as a reaction to the molecule absorbing light.
These new method was validated returning to the investigation of a couple of molecules previously studied by our group here: J. Phys. Chem. Lett. 2017, 8, 6148-6153. One of the molecules was emissive in crystal whilst the other was not, and we wished to recover these results. Additionally, the prediction of the previously available methods was for the molecule to shine blue, but experimental evidence said that it should be red.
The new ONIOM QM:QM’ methods managed to reproduce the correct behaviour of the two molecules, and recover the correct colour. We found that using QM:QM’ methods was important for this, as was extending the electrostatic interactions in the way described above. I implemented all of these methods into a program, which is described in a later publication.
-
Molecular and Crystalline Requirements for Solid State Fluorescence Exploiting Excited State Intramolecular Proton Transfer
Michael Dommett, Miguel Rivera, Rachel Crespo-Otero, J. Mater. Chem. C, 2020, 8, 2558-2568 , preprint
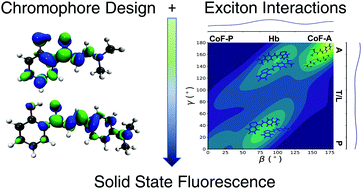
This was another great output by Michael. Essentially, he used the families of molecules from J. Phys. Chem. Lett. 2017, 8, 6148-6153, only including even more species, and a neighbouring family, in order to have enough diverse results to start extracting statistical conclusions. The focus remained on molecules used in lasers as crystals, which had a hydrogen atom transferring across the molecule upon absorption of a photon.
I don’t want to go too far in depth on this one, but what is most valuable here, in my opinion, is the development of a systematic way of approaching the modelling of these systems. The aim was to be very comprehensive, so as to weigh and compare all of the factors which could determine the emission of light in these materials, or the absence thereof. By repeating this very broad and time-consuming analysis for all-in-all eleven molecules, Michael developed a workflow which we have since then used as a template for investigating new chemistries.
The outcome was the formulation of a set of requirements for these proton transfer molecular crystals to be efficiently luminescent (very bright). We found that encouraging the transfer of the proton (i.e. hydrogen) was a strong contributing factor. Making the molecules stack in parallel arrangements was unfavourable for emission, but the transfer of the proton would usually trump this. Finally, making more rigid molecules would help encourage the emission of light, by discouraging the motions of the molecule which would allow it to lose its energy through heat and not photons.
In this paper, I developed a few tools to measure how tightly packed the molecules were in their crystal environment. I also assessed the importance of the interactions from very far ranging molecules within the crystal on the behaviour of the excited state. Finally, I handled a significant portion of the revision process.
-
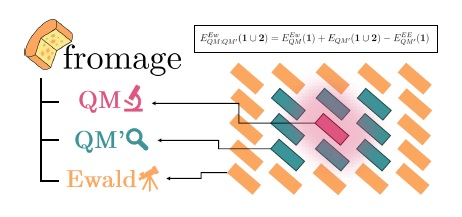
fromage: A library for the study of molecular crystal excited states at the aggregate scale
Miguel Rivera, Michael Dommett, Amir Sidat, Warda Rahim, Rachel Crespo‐Otero, J. Comput. Chem., 2020, preprint
This paper is maybe the most technical yet, but I made sure that all of the figures were very attractive, in order to not put off potential readers. This is an account of the program which compiles all of the tools developed during my PhD. fromage (FRamewOrk for Molecular AGgregate Excitations) is a Python library designed to assist in modelling excited states in molecular crystals. The need for it came about because, as discussed above, our perspective in this field encompasses molecules, clusters of molecules, and crystals, and these three orders of magnitude typically require different sets of tools to study. fromage offers ways of jumping between the three both for newcomers (who are supplied with readymade programs to use) and experts (who can use parts of fromage in their own short programs).
The bulk of the paper presents the readymade programs one by one, explaining how they work, showing how to use them, and concluding with an example for a real molecular crystal, relevant to applications to do with light. The series of programs had the following uses:
- measure the available space for a molecule inside a crystal to wiggle around
- detect neighbouring pairs of molecules and measure their conformations
- evaluate how much excited states involve neighbour molecules instead of just the one
- find how these excited states were shared between molecules
- calculate the excited states whilst taking into account their crystalline environment, as is described here: J. Chem. Theory Comput. 2019, 15, 2504-2516
In the end this paper is more of a good way of advertising the program, and also provides something to cite for future users. The real bulk of the work is in the source code and the documentation for fromage. Understanding the science is one thing, but it’s really the object of the program itself which I am proud of, since at the beginning I didn’t know any of the tools to make a sustainable program, a webpage, a tutorial, a cheese logo etc.
To find an up-to-date list, you may peruse my Google Scholar.